
Author: Shriti Kewat
Abstract:
Nearly 80% of the nitrogen expelled by humans and other mammals takes the form of urea, which is created by a sequence of events that take place in the cytosol and mitochondrial matrix of liver cells. The Krebs-Henseleit cycle or the urea cycle are the collective names for these processes. Ammonia is a harmful byproduct of the metabolism of nitrogen that needs to be eliminated from our bodies. In the mitochondria of liver cells, the urea cycle, also known as the ornithine cycle, transforms surplus ammonia into urea. After urea formation, the substance enters the bloodstream, passes through the kidneys’ filter, and is eventually eliminated as urine. The following is the general response when ammonia is converted to urea:
(Urea + Water + 3 ADP ——> 2 Ammonia + CO2 + 3ATP)
Introduction:
The series of events that produce urea and regenerate ornithine once arginine is generated from ornithine.
NH3 + CO2 + aspartate + 3 ATP + 2 H2O = urea + fumarate + 2 ADP + 2 phosphates + AMP + diphosphate is the overall reaction equation.
In a healthy adult, the elimination of ammonia as urea in the amount of 10 to 20 gm per day is mediated via the urea cycle. In severe situations, hyperammonemia encephalopathy and irreversible brain damage may arise from an incompletely functioning urea cycle. A hereditary deficiency, typically a congenital enzymopathy, or an acquired condition, like cirrhosis owning to alcoholism, can cause a failure of ureagenesis.
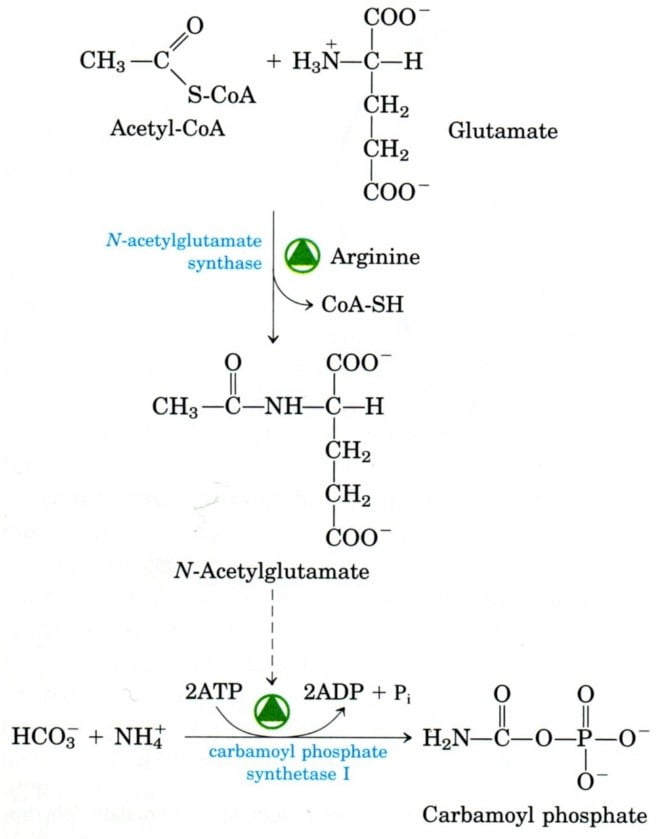
The urea cycle starts with two mitochondrial stages. The enzyme carbamoyl phosphate synthetase (CPS), located on human chromosome number 2, facilitates the conversion of NH3−, HCO3−, and ATP into carbamoyl phosphate. NAG synthetase converts glutamate and acetyl-CoA into N-acetyl glutamate (NAG) is a crucial ureagenesis regulator and a required CPS effector. Many factors increase the amount of NAG in mitochondria, such as arginine, dietary protein, and corticosteroids.
- Ammonia needs to be removed
The breakdown of amino acids, which is mostly caused by the breakdown of both dietary and intracellular proteins, is the source of ammonia: digestive enzymes, proteins that are produced during cell digestion sloughed off the GIT muscle walls. Intracellular haemoglobin proteins (damaged, unneeded)
- Ammonia has to be eliminated
As ammonia combines with α-ketoglutarate, it limits the TCA cycle and causes a drop in ATP levels, which makes it hazardous, particularly for the central nervous system. Elevated ammonia levels linked to metabolic diseases or liver damage can cause tremors, speech impediments, impaired vision, unconsciousness, and even death. Blood ammonia levels should be between 30 and 60 µM. Oxidation of amino acids and urea generation
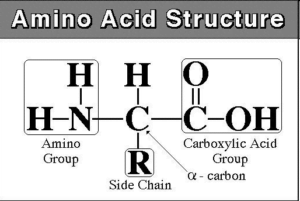
Elimination of nitrogen from amino acids:
- First, take off the amino group.
- Step 2: Transport the amino group to the liver for the elimination of nitrogen.
- Step 3: Mitochondrial entry
- Step 4: Get nitrogen ready for the urea cycle.
- Step 5: Cycle of urea
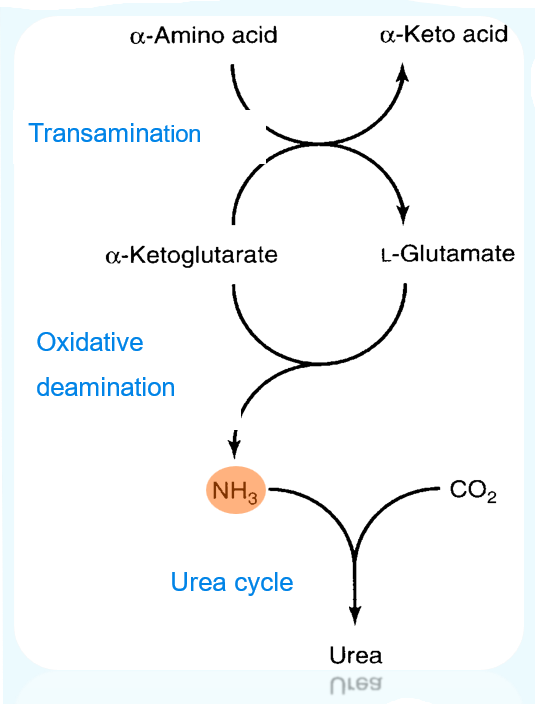
- Nitrogen in excretory forms
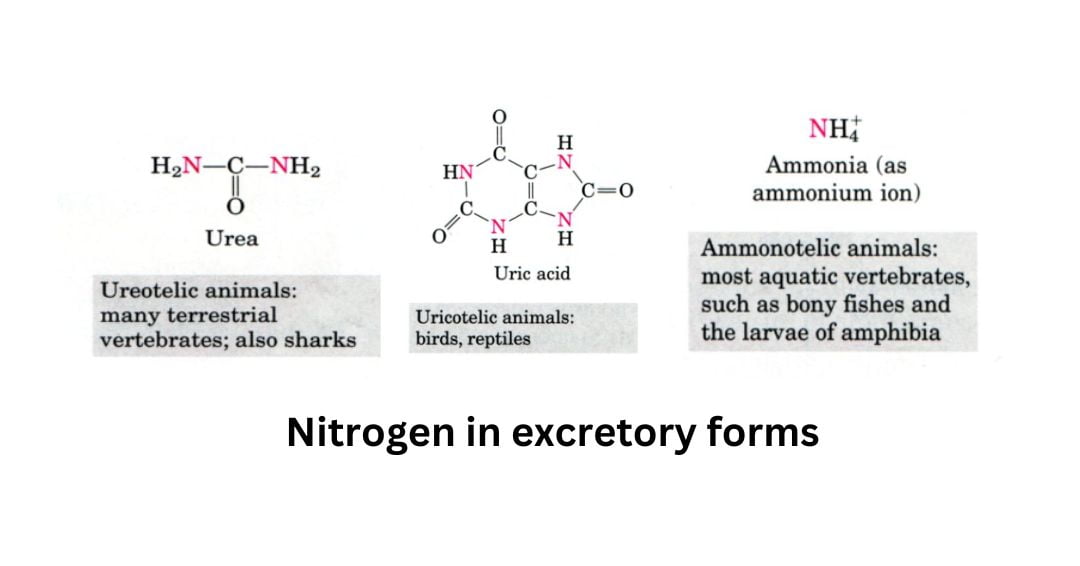
- Excess NH4+ is expelled as uric acid (found in birds and terrestrial reptiles)
- urea (found in many terrestrial vertebrates)
- ammonia (found in microorganisms, aquatic vertebrates, or amphibian larvae).
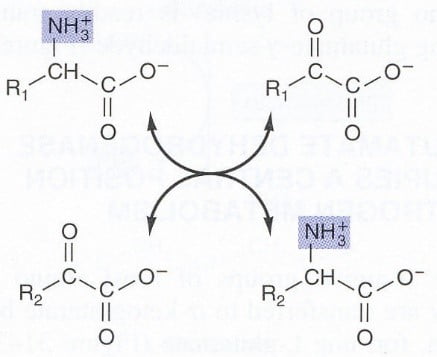
Step 1. Remove amino group
- Transferring an amino acid amino group to a -keto acid results in the conversion of the original AA to the corresponding -keto acid and vice versa:
Step 2: Take an amino group to the liver for nitrogen excretion
- In the liver, glutamate releases its amino group as ammonia.
- The amino groups of L-glutamate molecules, which are derived from numerous a-amino acids, are accumulated in the liver.
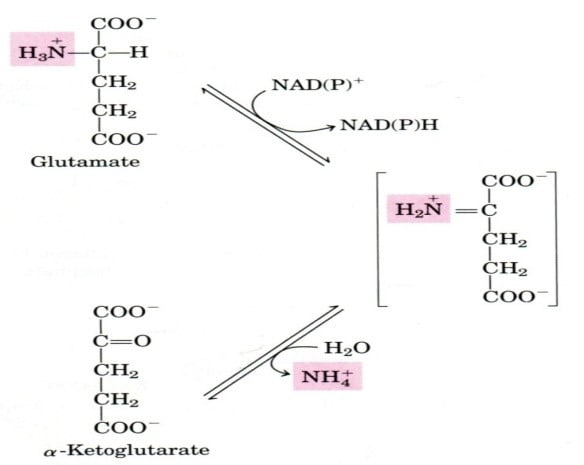
Step 3: nitrogen enters the mitochondria Carriers of nitrogen:
alanine, glutamine, and glutamate
Two enzymes within the liver and two outside the liver:
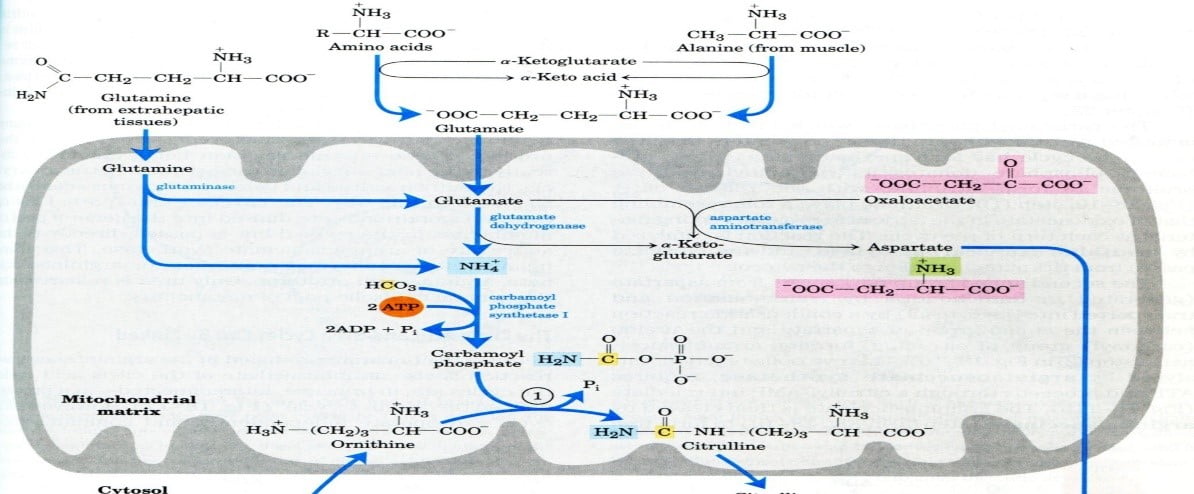
PLP → a-ketoglutarate → glutamate via aminotransferase
Step 4: get nitrogen ready for the
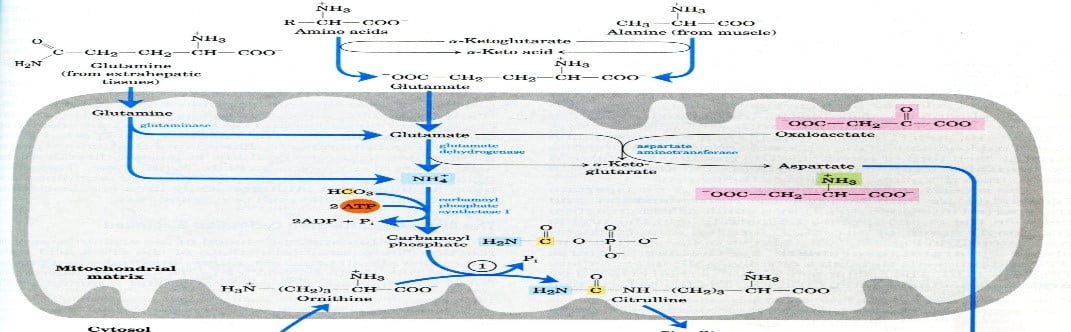
Step 5: Cycle of urea
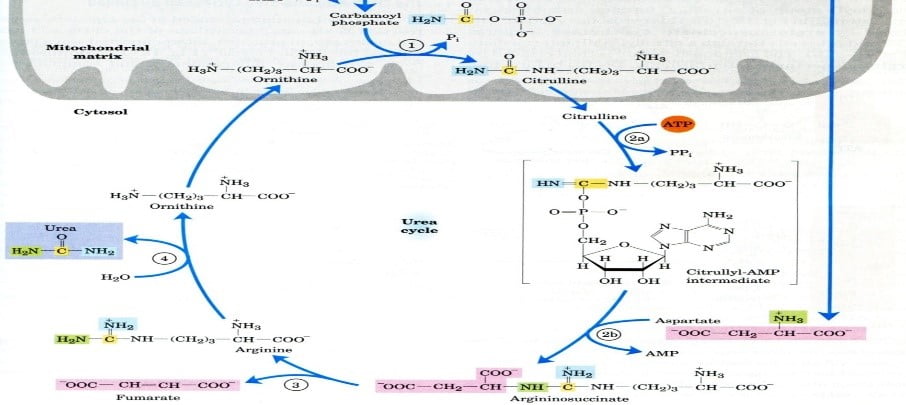
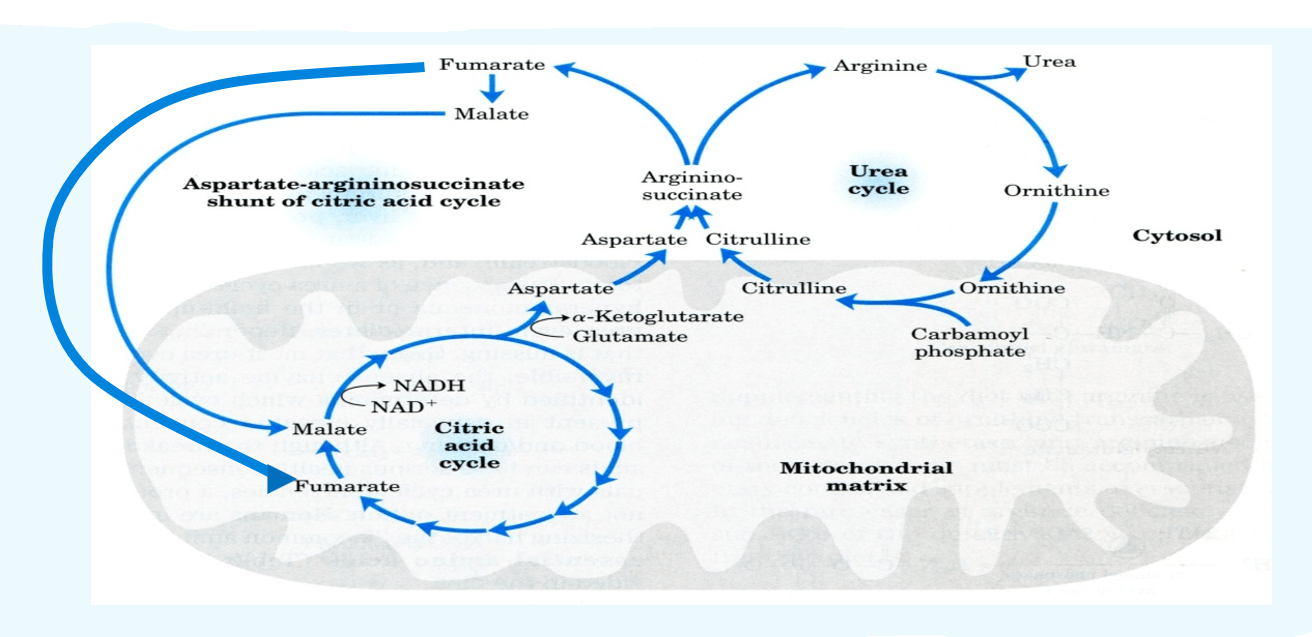
The review of the urea cycle (reaction sequence)
- The urea cycle requires the production of carbamoyl phosphate in mitochondria (Carbamoyl phosphate synthetase).
- Ornithine (Ornithine transcarboxylase) and carbamoyl phosphate combine to generate citrulline.
- The extra nitrogen needed to create arginosuccinate in the cytosol is supplied by aspartate (Arginosuccinate synthase).
- Fumarate and arginine synthesis (arginosuccinate lyase)
- Arginine is hydrolyzed by arginase to produce urea and ornithine.
The overall chemical equilibrium of urea biosynthesis
- NH3 + CO2 + 2ATP → carbamoyl phosphate + 2ADP + Pi
- Carbamoyl phosphate + ornithine → citrulline + Pi
- Citrulline + ATP + aspartate → argininosuccinate + AMP + PPi
- Argininosuccinate → arginine + fumarate
- Arginine → urea + ornithine
- Sum: 2NH3 + CO2 + 3ATP g urea + 2ADP + AMP + PPi + 2Pi
Management of the urea cycle
- Two stages govern the urea cycle’s activity:
- Protein makes up the majority of the diet, along with a lot of urea (amino acids are used as fuel).
- It extended fasting results in the breakdown of muscle proteins and high levels of urea.
- Animals manufacture enzymes more quickly when they are starved or fed an extremely high-protein diet.
- In well-fed animals that consume a diet high in fat and carbohydrates but low in protein, the rates of enzyme synthesis are reduced.
- Variations in the demand for urea cycle activity control the rate of synthesis of four urea cycle enzymes and carbamoyl phosphate synthetase I (CPS-I) in the liver.
- An equal amount of nitrogen excreted balances the amount of nitrogen consumed.
- Urea makes up about 80% of nitrogen excreted in the body.
- The rate of synthesis of four urea cycle enzymes and carbamoyl phosphate synthetase I (CPS-I) in the liver is regulated by changes in demand for urea cycle activity.
Enzyme abnormalities in the urea cycle
Ammonia toxicity – Elevated ammonia content in blood and other bodily fluids causes ammonia to permeate into cells and penetrate the blood-brain barrier, which in turn increases the production of glutamine and glutamate from a-ketoglutarate. Depletion of a-ketoglutarate in the brain results in suppression of the TCA cycle and ATP generation. The CNS effects, such as strange behaviour, may be attributed to the neurotransmitters glutamate (an excitatory neurotransmitter) and GABA (an inhibitory neurotransmitter).
- A new-born completely devoid of one or more enzymes survives for a few days at the very least.
- A lot of partial defects in enzymes result in changed Km values.
- There are cases of known enzyme deficits.
- Each point in the cycle where it was broken had a varied effect on nitrogen metabolism; some of the intermediates could diffuse from hepatocytes, collect in the circulation, and then move into the urine.
- If signs are not identified promptly, they can lead to severe mental impairment and irreparable brain damage.
- N-acetyl glutamate synthase deficiency:
Urea cycle failure can result from an autosomal recessive enzyme deficiency or genetic mutation.
if not identified right away after delivery, a serious new-born illness could be fatal,
neonate with hyperammonaemia and overall hyper aminoacidemia (liver not able to manufacture N-acetyl glutamate).
Weakness, vomiting, and a deep coma are among the initial signs. Treatment with N-carbamoyl-L-glutamate, a structural analogue, activates CPS-I, lessens the disorder’s severity,
- Carbamoyl phosphate synthetase (CPS I) deficiency:
metabolic disease that is autosomal recessive and linked to developmental delay and mental disability.
Between 0 and 50% of the normal amount of CPS-I production in the liver, hyperammonemia has been reported.
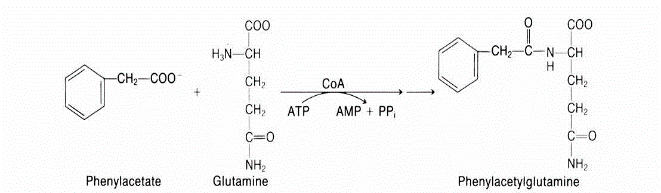
- Deficiency in ornithine transcarboxylase (OTC)
The most prevalent urea cycle problem, causes the enzyme to mutate and become inefficient.
Males are typically more severely affected than females in this X-linked recessive condition, which is caused by multiple distinct mutations. Developmental delay and mental impairment are two possible side effects of OTC use.
Urine containing Hippurate and Phe-Ac-Gln is discharged after treatment with benzoate and phenylacetate.
- Arginosuccinate lyase deficiency (arginosuccinate aciduria)
A rare autosomal recessive condition known as arginosuccinate aciduria (arginosuccinate lyase deficiency) causes excessive arginosuccinate excretion in the urine.
The degree of symptoms varies widely, making it challenging to assess the effectiveness of
- Arginosuccinate synthase deficiency – citrullinemia (citrullinemia)
Citrullinemia, also known as citrullinemia, is an autosomal recessive metabolic condition caused by arginosuccinate synthase insufficiency, or the inability to condense citrulline with aspartate.
urine elimination and blood accumulation of citrulline.
The first few days of life are typically when type I citrullinemia is first noticeable.
The signs and symptoms of type II citrullinemia, which primarily affects the neurological system, typically manifest in adulthood.
Therapy: targeted arginine supplementation to promote protein synthesis and the production of ornithine and creatin treatment; dietary limitation of nitrogen is helpful in this regard.
- Arginase deficiency (arginine Mia)
Extremely rare autosomal recessive condition causing several problems in CNS function and development. urine’s ability to accumulate and excrete arginine as well as its precursors products during metabolism. Low-nitrogen diet therapy, which includes critical amino acids
In conclusion
The liver uses the urea cycle, a sequence of metabolic processes, to change ammonia into urea. Six enzymes make up the urea cycle, which helps the body eliminate nitrogen generated during the metabolism of amino acids. It is broken down into urea and expelled in the urine.
Carbamate, another name for urea, is a chemical with a long history that is safe and beneficial. It is a naturally occurring substance that is widely present in the urine of mammals and is created by the metabolism of proteins. Hepatocytes, or liver cells, have mitochondria where the urea cycle begins, and the cytoplasm is where the cycle ends. After that, the finished product is moved to the kidney, where the body excretes it. Ammonia is produced when amino acids are broken down or when nitrogen is metabolized. Extra ammonia is eliminated by the Krebs-Henseleit Cycle or the Urea Cycle. The liver produces urea, which is then expelled in the urine. Thus, urea is the final byproduct of mammalian nitrogen metabolism.
References
- Singh, R. H., Rhead, W. J., Smith, W., Lee, B., King, L. S., and Summar, M. Nutritional management of urea cycle disorders’ Care Clin.21:Pages: S27-S35. 2005.PM:16227113
- National Urea Cycle Disorders Foundation (information, referrals, resources, patient/family education and support)
- L. Nelson, M. M. Cox : LEHNINGER. PRINCIPLES OF BIOCHEMISTRY Fifth edition